
钜大LARGE | 点击量:4811次 | 2018年08月03日





为何存在电池电压衰减的情况
研究亮点:
电压衰减的根源为失氧
失氧造成所有过渡金属的平均价态不断变低(Ni3+/Ni4+电对转化为Ni2+/Ni3+电对,Mn4+/Mn5+电对转化为Mn3+/Mn4+电对,Co3+/Co4+电对转化为Co2+/Co3+电对),导致电压衰减
失氧还会造成材料颗粒的微瑕疵(如在颗粒内部形成大孔),导致电压衰减
技术亮点:
同步X射线吸收光谱(XAS)具有加大的穿透功率,能穿透整个阴极,提供体相分析
STEM具有原子层面的空间分辨率,提供表层分析(5nm)
新型三维电子断层成像技术,观察孔隙在三维空间上的生长
电压衰减已经成为阻止一系列高能量密度电池电极商业化的重要原因。富锂材料,得益于较高的容量(>250mAhg-1vs.<200mAhg-1),是替代传统阴极材料的理想选择。但其在循环过程中,平均电压不断衰减,严重影响能量效率且给电池管理系统带来困难。电压为何衰减?近日,布鲁克尔海文国家实验室联合中科院物理所和阿贡国家实验室,使用X射线光谱和三维电子显微镜成像技术,原位和非原位探索材料在充放电过程中,体相和表面的变化。
氧化还原电对的演变
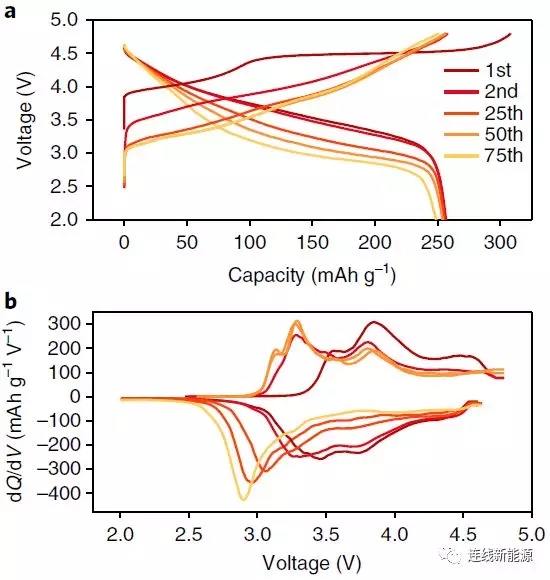
图1Li1.2Ni0.15Co0.1Mn0.55O2的电化学性:a.充放电曲线;b.dQ/dV图。电压衰减问题显而易见
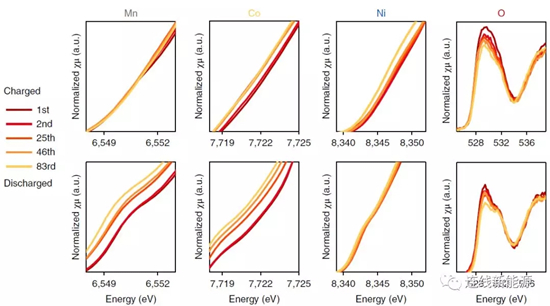
图2Li1.2Ni0.15Co0.1Mn0.55O2中不同元素的XAS结果。随循环进行,三种金属的平均价态持续降低,证明体相中过渡金属和氧的配位能力在减弱。
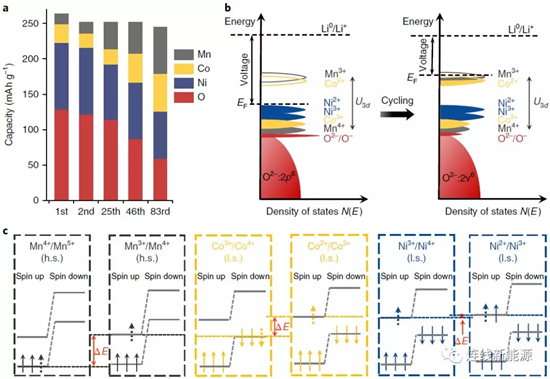
图3Li1.2Ni0.15Co0.1Mn0.55O2在循环过充中,氧化还原电对的演化:a.每种元素对放电容量的贡献;b.费米能级由于电子结构的变化而升高;c.不同氧化还原电对得失电子时涉及到的不同能级
如图3a,首周时,O和Ni对容量贡献占主导,然而,随循环进行,Mn和Co对容量贡献增多,逐渐补偿O和Ni对容量贡献的减少,最后Co和Mn的容量占主导地位。氧化还原电对从O和Ni逐渐转变成Mn和Co,势必会影响电压分布。如图3b,阴极材料的能态密度在不断改变。电池的开路电压(OCV)由费米能级相对于Li+/Li0能级的差别决定,与将电子从富锂阴极移动到Li阳极的功焓有关。起初,材料的费米能级位于Ni2+/Ni3+氧化还原电对之上,循环后,发生氧气释放并导致TM还原。如Ni倾向于首先在表面还原,形成电子绝缘的非活性岩盐相,降低Ni对容量的贡献;Mn和Co的还原产生Mn3+/Mn4+和Co2+/Co3+氧化还原电对,这种还原能将费米能级变得更高,导致OCV变低以及工作电压变低。这个过程也能解释为什么O对容量的贡献逐渐减少,由于TM还原,TM与O的共价键减弱,造成参与氧化还原的O减少。
比较图3c不难发现,Ni2+/Ni3+与Ni3+/Ni4+电对的能级差最小(△E),而Co2+/Co3+和Co3+/Co4+电对的能级差加大,Mn3+/Mn4+与Mn4+/Mn5+电对的能级差更大。能级差的不同是因为电子结构不同。对Mn和Co而言,从一个电对变到另一个,需要设计不同的轨道。如Co2+/Co3+电对变成Co3+/Co4+电对,涉及到在自旋向上eg轨道上失去(氧化)或得到(还原)一个电子。然而对于Ni,Ni2+/Ni3+电对与Ni3+/Ni4+电对的转变,涉及的电子得失是在同一轨道(自旋向上eg)。因此,Mn和Co是OCV下降和所谓电压衰减主要原因。
表面反应
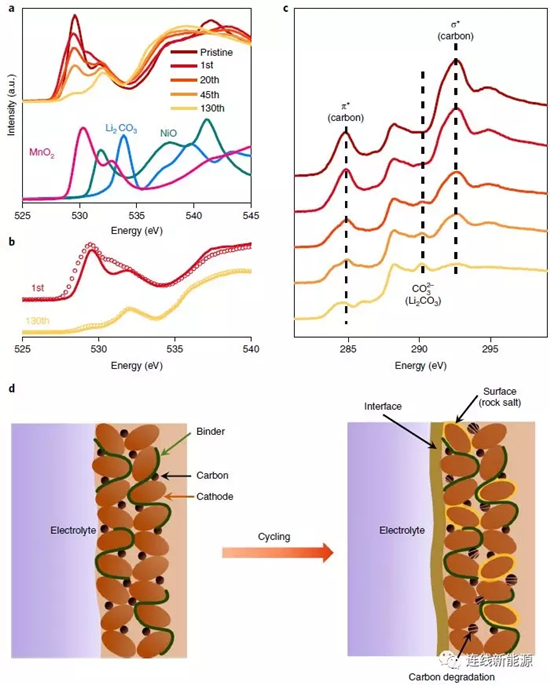
图4Li1.2Ni0.15Co0.1Mn0.55O2表层分解导致过电位增大:a.富锂材料在充电态下的O谱及各参考组分的O谱;b.充电态(圈)与放电态(线)的O谱;c.C谱;d.循环前后的示意图,循环后形成新界面、表面发生重组且导电碳降解
前面所述的XAS能提供丰富的体相反应信息,而软XAS技术则能对材料的表面进行表征(5nm)。O谱所有的峰如图4a所示,随循环进行,代表MnO2的峰不断减弱,而代表NiO的特征峰不断增强,证明表面结构发生重组,岩盐结构因此生成。图4b中代表Li2CO3的峰(535eV以上)逐渐变强,代表表面OH物种增多,是因为电解液分解而形成不同的有机/无机化合物,如Li2CO3、RCO2Li等。C谱所有的峰如图4c所示,284.8eV和292.6eV归结于导电碳的π反成键和σ反成键,290.2eV归结于Li2CO3中的CO32-。随循环进行,导电碳的特征峰减弱,证明导电碳黑逐渐分解,可能是因为在高电压下,PF6-嵌入石墨中。相反,归属于Li2CO3的峰增强,Li2CO3代表阴极界面膜(CEI)的产物,意味着CEI随循环进行在不断生长。CEI的增长会使电化学反应动力学更加滞后,造成过电势增加且电压衰减。
微结构瓦解的开始和蔓延
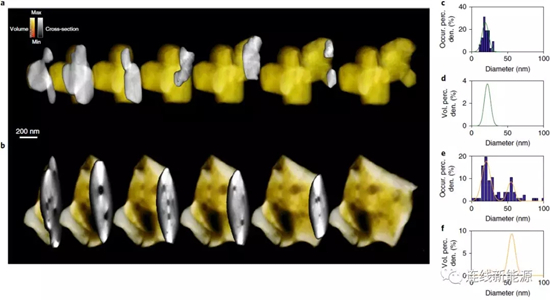
图5Li1.2Ni0.15Co0.1Mn0.55O2材料的三维断层图:a.初始态和b.循环15周后的颗粒截面图;内部孔径分布图(c、d)初始态和(e、f)循环15周后
失氧怎样诱使材料的微观结构发生改变?研究人员采用ADF-STEM成像和空间分辨EELS来研究结构的成核和演变过程。初始态和循环15周后,阴极材料的3D内部结构如图5a、b所示,图5b能观察到循环后颗粒内部产生一些大孔。通过定量分析孔径,可推断这些大孔在一开始并不存在(图5c、d),但15周时,大孔占据材料孔隙率的绝大部分(图5e、f)。这种大孔是在循环的过程中产生的,很可能是由失氧所留下的成核空位演化而成的。
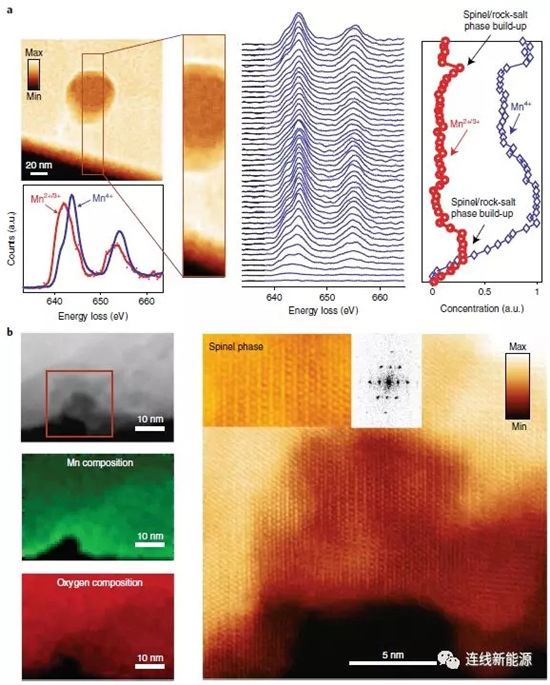
图6颗粒表面暴露的孔隙(a)和体相隐藏的孔隙(b)的空间分辨EELS分布
为进一步弄清这些大孔的成因,科研人员采用STEM-EELS技术来分析材料颗粒中的孔结构,例如,研究对象为颗粒体相中一个隐藏的孔隙和颗粒表面一个暴露的孔隙。图6a显示隐藏的孔周围形成了一层薄的Mn2+壳。由于这些小孔隙发现于初始材料中,故形成于材料的制备过程,其形状和大小保持不变直至在其周围开始产生氧气(循环过程中)。与之对应的是暴露的孔隙,这些孔隙与电解液接触,会逐渐形成一层厚的尖晶石相/岩盐相,因此,EELS相对浓度分布表明,表面体积中的Mn相对含量增加。其实,很多孔隙既不是完全隐藏也不是完全暴露的。这种部分暴露的孔隙周围,存在氧气扩散通道(以微结构瑕疵、位错和晶界的形式存在),随循环进行,孔会变多且变大,与那些完全暴露的孔隙一起,加速结构的相变、O2的进一步释放、微结构瑕疵的产生和电压衰减。
如何抑制失氧
失氧是电压衰减的根源,怎样才能抑制失氧?一种方法是对材料表/界面进行处理。例如,表面包覆能减少电解液与界面的直接接触。注意最好对初级颗粒进行包覆,而非次级颗粒。除包覆外,还可以在颗粒表面引入氧空位,这种表面处理方式能显著减少失氧。另一种方法就是体相掺杂。例如,掺杂Al阳离子,Al能占据四面体位点,因此能抑制过渡金属离子向四面体空位迁移,这种迁移过程是伴随相转变过程与失氧过程同时发生的。
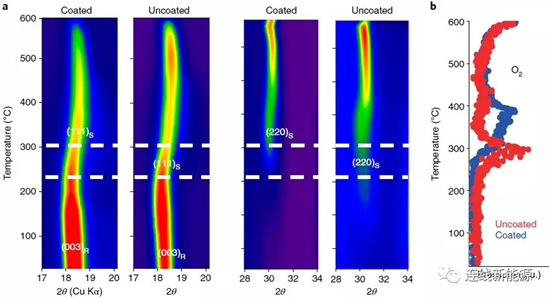
图7Li1.2Ni0.15Co0.1Mn0.55O2材料与经AlF3包覆后的热稳定性对比:a.加热过程中的原位XRD说明没包覆的材料更快经历相变(R代表斜方六面体相,S代表尖晶石相);b.加热过程中的氧释放情况,没包覆的材料在更低的温度下就会释放氧。
无论测试循环性能测试还是热稳定性测试,都涉及氧的释放和随之引起的过渡金属的还原、迁移。相比之下,热稳定性测试的耗时短、用量少,因此可以替代循环测试,评估材料的结构稳定性。材料热稳定性的高低可直接用来评估电压衰减的程度。
结论
失氧是引起电压衰减问题的根源。氧释放会造成材料更高的比表面以及使颗粒内部产生更多的缺陷,这将进一步加速氧释放过程;氧释放引起Mn和Co的还原(向低价态电对Mn3+/Mn4+和Co2+/Co3+转化),造成电压衰减;此外,氧释放加速表面重组,阴极界面膜(CEI)的传质变慢且电化学性能变差,造成电压衰减。今后的科研工作应围绕抑制氧释放展开,主要途径为表/界面处理或体相掺杂。
上一篇:让快充更加安全的新型锂电池材料
下一篇:动力电池业两大新规今起实施


















